Anton 2 Simulations Kick off Drug Safety Prediction Pipeline
Many possibly useful drugs are abandoned because they increase the QT interval of the heartbeat. This is because QT prolongation can cause life-threatening side effects. But not all QT-prolonging drugs are toxic. Using the D. E. Shaw Research Anton 2 supercomputer hosted by PSC, scientists from the University of California, Davis, and the University of Calgary kicked off a “pipeline” of simulations that enabled them to predict which QT-prolonging drugs may be safe enough to continue testing. This pipeline could be applicable to any drug, enabling companies to abandon ones that will cause serious side effects faster and to keep developing ones that won’t.
Why It’s Important
Many useful drugs—including common antimicrobials such as azithromycin and hydroxychloroquine, antidepressants and antipsychotics—can cause serious side effects of on the heart. The most common form of such cardiotoxicity, called drug-induced QT prolongation, stretches out a critical part of the heartbeat. Severe QT prolongation can cause a number of fatal effects.
“One of the most common health complications for many therapeutics of different classes—antivirals, antinflammatories and others—is that they may cause palpitation and irregular heartbeat. In many cases, this can represent significant cardiotoxic risks. These drug-induced changes in heart electrical signaling are often related to interactions with one of the cardiac channels known as hERG1. In the pharmaceutical industry right now and many municipalities around the world, every drug has to be tested for interactions with this channel in pre-clinical development before moving to clinical trials.”—Colleen E. Clancy, University of California, Davis
The QT-prolonging drugs all act by binding to a heart-muscle-cell protein called hERG1. This protein is a channel in the heart-cell membrane that normally opens to let potassium ions cross the membrane at the end of a heartbeat. This helps the heart “reset” for the next beat. But there’s a puzzling difference between drugs blocking hERG1 ion currents. Some can be very toxic to the heart. But others are not. Many promising drugs that manufacturers abandon because they lengthen the QT interval might turn out to be safe, if scientists could predict which ones are which.
A team of scientists led by Colleen Clancy and Igor Vorobyov at the University of California, Davis, and Sergei Noskov at the University of Calgary in Canada built a series of computer simulations to make such predictions. Their simulations span from the activities of atoms to molecules to cells to tissues. This massive computational pipeline required simulations on a number of computer systems.
But the pipeline had a problem at step one. Nobody knew how the drugs entered the heart cells and attached to hERG1 in the first place. Replicating these events required unusually long computer simulations. The scientists needed the Anton 2 supercomputer, which was developed and made available without cost for non-commercial research by D. E. Shaw Research, and is hosted by PSC with support from the National Institutes of Health.

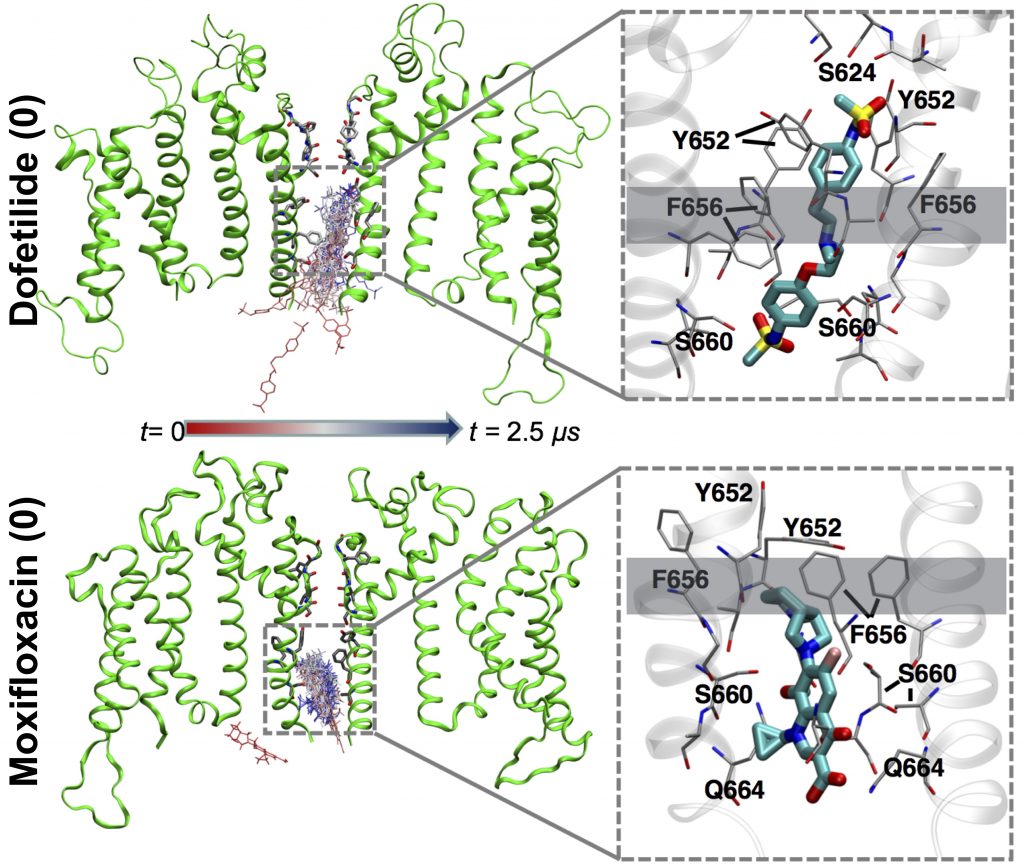
Dofetilide (top) binds to the heart’s hERG1 membrane channel when its central pore is closed, preventing it from working entirely and causing serious heart-rhythm side effects. Moxifloxacin (bottom) binds to the open channel, allowing it to partially work and not causing those side effects.
The team compared simulations of two QT-prolonging drugs: the cardiotoxic irregular-heartbeat drug dofetilide, and the nontoxic antibiotic moxifloxacin. Their initial simulations on Anton 2 began with drug molecules in the aqueous solution outside a simulated cell membrane containing hERG1 protein.
“We were really in the dark as to how the drug accessed the cell. We needed to simulate a lot of drug molecules just to see where they would go. We have a lot of [computing] resources locally, but for a system of that size … [using] a standard GPU or CPU platform, we probably would still be waiting for a single event. Anton 2 simulated something like 5 to 7 microseconds a day, which made the work possible. PSC staff has been of tremendous help to promptly identify and solve any issues with simulation setup, performance and analysis.”—Igor Vorobyov, University of California, Davis
To reproduce these systems, the scientists had to simulate some 130,000 to 240,000 atoms contained in drugs, protein, water, ions, and the fatty lipid molecules that make up cell membranes. And they needed to run what amounts to a very long simulation—around 2.5 microseconds of simulated time for an individual run or 5 to 10 microseconds for each drug counting several trials and drug ionization forms. That’s only 5 to 10 millionths of a second, but in computational time it’s huge, totaling billions of discrete simulation time steps. More, to mimic the activity of the hERG1 channel to determine its functional state in drug binding simulations, they needed to wait for potassium ions to start to move through the channel. Ultimately, this took 10 to 20 simulated microseconds in total considering different salt concentrations, simulation and model parameters. Anton 2, which is specially built to perform very long molecular dynamics simulations, was crucial for the job.
In the simulations, both drugs crossed the membrane by shouldering through the lipid molecules that make it up and were observed to spontaneously move into an open channel pore on the intracellular site. But the two differed after this. Potentially cardiotoxic dofetilide bound to the channel more tightly when it was in a non-conducting, so called inactivated state, keeping the channel pore closed and unable to work. Moxifloxacin bound to the open channel, allowing it to partially function. This prediction agreed with previous lab results, giving the scientists confidence their simulation was on target.
“About five years ago, an experimental finding showed it was possible that what’s a really important factor in determining drug cardiotoxic profiles is not binding to the channel, but binding to a specific state of this membrane protein. If the drug binds to the inactivated state it locks the pore into the closed state, which is far more damaging … Our models turned out to be fairly predictive, with binding to the inactivated state one of the major determinants of cardiotoxicity.”—Sergei Noskov, University of Calgary
Better, the Anton 2 simulations set up the team’s other simulations to work properly. Starting with the movements of individual drug molecules floating around the hERG1 channel, validating its structural state from published Cryo-EM studies and then developing models for drug binding to various channel states and proceeding to cell and tissue behavior, each step accurately recreated the effects of the drugs on cardiac tissue activity. The pipeline of simulations could potentially be used to test any drug for cardiotoxicity, speeding acceptance of safe drugs and helping doctors rule out unsafe ones faster as well.
These studies have been generously funded by the National Institutes of Health, Canadian Institutes of Health Research, American Heart Association and, in addition to using the Anton 2 at PSC, have been using supercomputing resources through Compute Canada, The Extreme Science and Engineering Discovery Environment (XSEDE), National Center for Supercomputing Applications (NCSA) Blue Waters, and Texas Advanced Computing Center (TACC) Frontera.
The scientists reported their results in two journal papers, one in the Proceedings of the National Academy of Sciences in February 2020 that you can read here, and one in Circulation Research in April 2020 that you can read here.